Updates on Modeling the Vascular Basement Membrane in the μSiM-hNVU
Introduction
The blood-brain barrier (BBB) plays an important role in maintaining brain chemistry by restricting the permeability of small molecules and cells. Inflammatory diseases may disrupt the BBB and promote the development of neurological disorders [1]. One potential mechanism of BBB disruption is the degradation of the extracellular matrix proteins that make up the endothelial basement membrane (BM). The BM is composed primarily of collagen IV and laminin networks that are linked by nidogen, perlecan, and heparan sulfate proteoglycans [2]. These proteins provide a support structure for endothelial cells, and it is believed that cellular adhesion to collagen IV is part of a pathway responsible for upregulation of the tight junction protein claudin-5 [3]. The tight junctions are the key to limiting endothelial permeability, since they restrict paracellular migration of small molecules. Thus, the BM helps maintain the barrier function of the BBB in two ways: its networks act as physical impediments to molecular diffusion, and they help sustain tight junctions. BM proteins are degraded by matrix metalloproteinase-9 (MMP-9, also known as Type IV Collagenase or Gelatinase B), which is upregulated by pro-inflammatory signals such as tumor necrosis factor-α (TNF-α) [4]. This could be an important link between inflammation and BBB disruption. Our goal is to model this effect using the μSiM-hNVU.
This post summarizes efforts made in the Gaborski NanoBio Materials Lab during the Spring 2022 semester. The majority of this work has been performed by co-culturing two cell types: a human cerebral microvascular endothelial cell line (hCMEC/D3; Sigma-Aldrich cat. no. SCC066) and primary human brain vascular pericytes (HBVPs; ScienCell cat. no. 1200). Co-culturing is accomplished by coating the trench side of the μSiM membrane with 2 μg/cm2 poly-L-lysine prior to “trench-down” device assembly, followed by inverted seeding of the HBVPs. hCMEC/D3s are seeded in the 25 µg/cm2 collagen I + 5 µg/cm2 fibronectin-coated top well 1-2 days later. Devices are subsequently maintained for 6-8 days prior to starting experiments to ensure that there is sufficient time for BM protein deposition.
Results
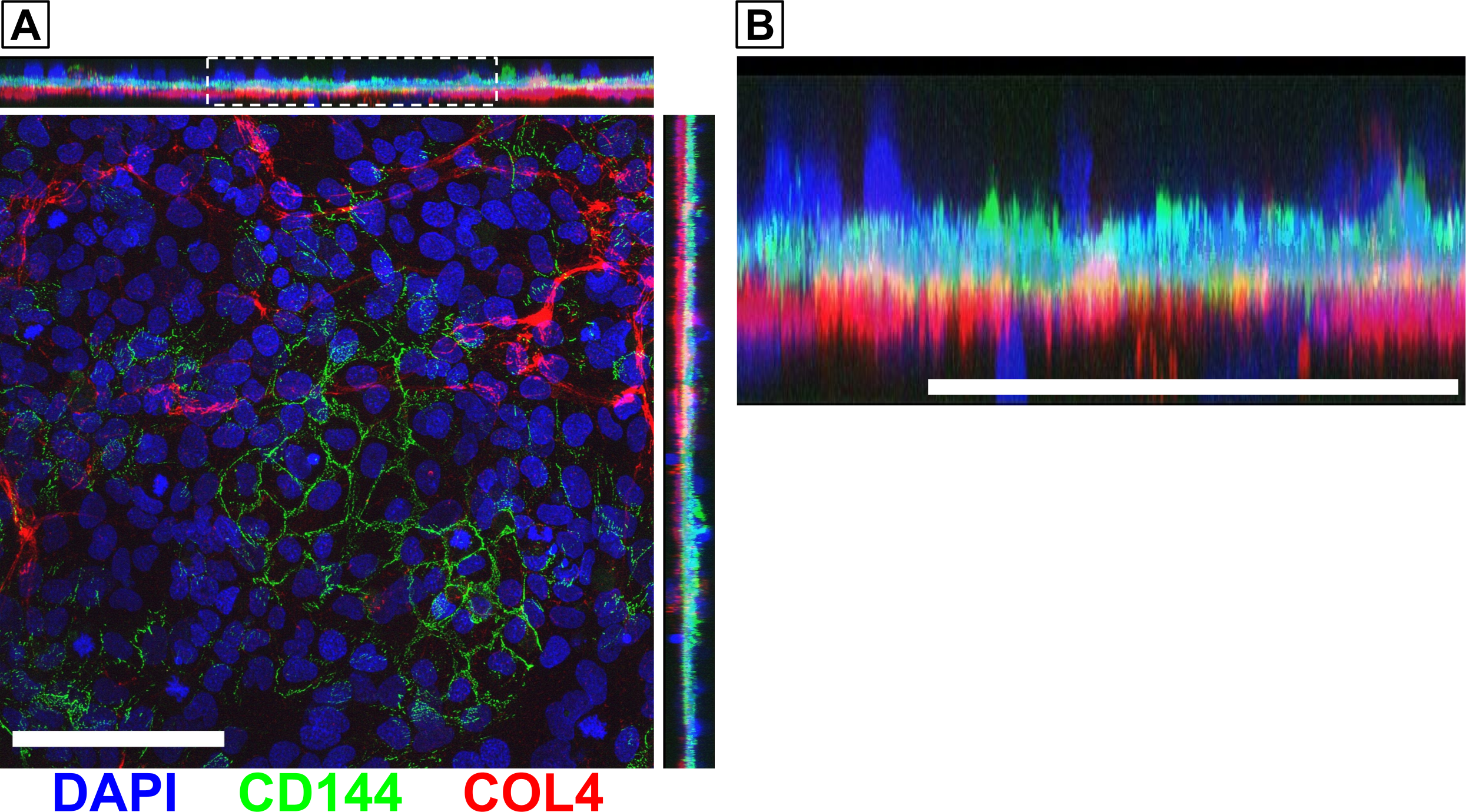
In Fall 2021, we were able to obtain high quality confocal microscopy images demonstrating that there was a continuous layer of collagen IV located below a continuous cell layer. While this provided good evidence that the endothelial cells were located above the BM (mimicking the in vivo orientation), we wanted to perform a follow up experiment to demonstrate this with more certainty. Newer images captured on the Andor Dragonfly Spinning Disc Confocal Microscope show a layer of endothelial-specific VE-cadherin above the collagen IV layer (Fig. 1). Other cells seem to be embedded within or located below the BM. These latter cells must be the pericytes since they lack VE-cadherin expression.
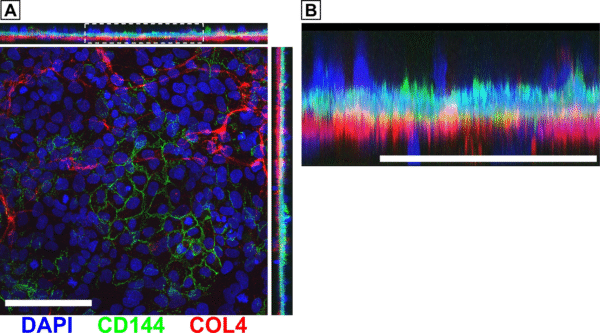
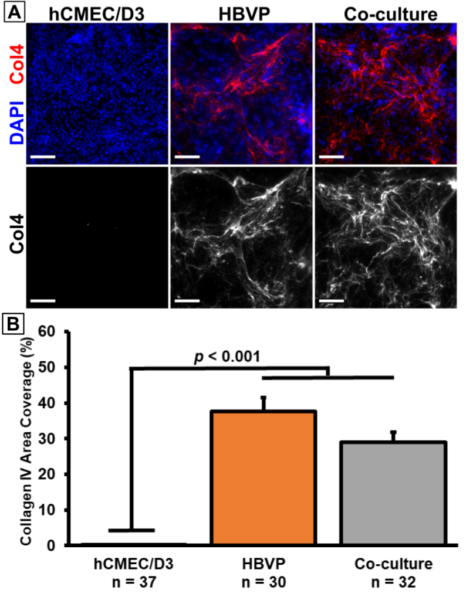
We also took confocal images of hCMEC/D3 and HBVP monocultures. The hCMEC/D3 monocultures showed VE-cadherin, but not much collagen IV (Fig. 2A). Conversely, the HBVP monocultures showed collagen IV, but no VE-cadherin (Fig. 2B). This matches our expectations based on prior results demonstrating that HBVPs are likely the main contributors of collagen IV in our BBB model. In fact, we have created a large dataset (n >30) of monoculture and co-culture device images that confirmed this finding (Fig. 3).
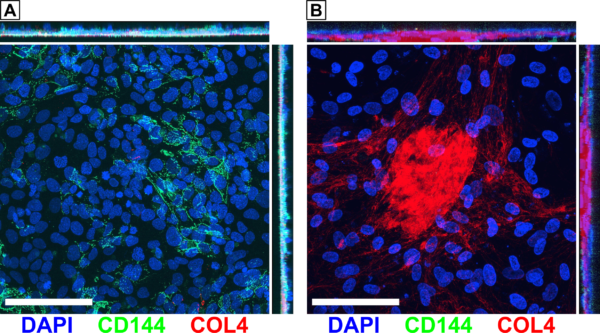
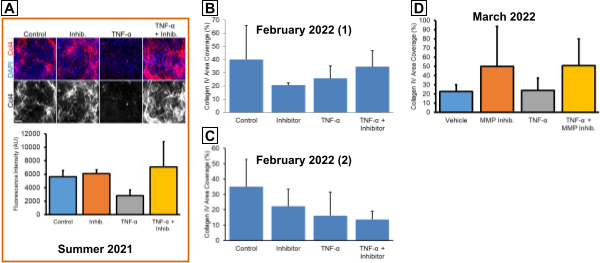
One important interaction that we wanted to model is the degradation of the BM when the BBB is exposed to inflammatory conditions. We previously observed that 24 hr treatment with 20 ng/ml TNF-α resulted in a loss of collagen IV, while co-treatment with 100 µg/mL MMP-2/MMP-9 Inhibitor I was able to rescue the collagen IV (Fig. 4A). This was promising, but we wanted to repeat this experiment to increase our limited sample size. However, multiple attempts have demonstrated poor repeatability since TNF-α treatment tended to not decrease collagen IV coverage compared to the vehicle control. In the first such attempt, treatment with the MMP inhibitor alone appeared to decrease collagen IV coverage (Fig. 4B). The second attempt seemed to show decreased collagen IV in all conditions compared to the control. In the third attempt, the inhibitor treatments increased collagen IV area coverage above the level found in the vehicle controls (Fig. 4D). In all cases, the results featured large standard deviations for some conditions. These experiments may indicate an issue with our stock TNF-α and MMP inhibitor, which were first stored in our lab’s -20°C freezer in October 2020. The TNF-α definitely seemed to be active upon initial receipt since we observed the prior BM degradation as well as dose-dependent responses in migration experiments. However, it is possible that this cytokine has lost activity over the 1.5 yr storage period. This may be equally true for the MMP inhibitor.
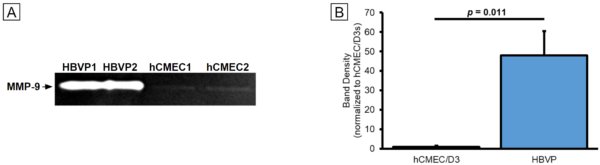
In addition to recreating inflammatory responses in our µSiM co-cultures, we sought to demonstrate that TNF-α does, in fact, cause increased MMP-9 activity. We also wanted to determine which cells were the primary sources of MMPs in our model. Monocultures of hCMEC/D3s and HBVPs were maintained in 6-well plates for 1 week prior to a 24 hr period in serum-free media. Conditioned media was collected and frozen at -80°C. This media was subsequently thawed and run through zymography gels. MMPs were renatured and allowed to digest the gel, producing clear bands with densities reflecting the activity levels of the enzymes. Bands were observed in all of our samples (Fig. 5A). HBVPs produced much more MMP-9 activity compared to the hCMEC/D3s per equal volume of cell culture media (Fig. 5B). This may indicate that HBVPs are much more involved in the dynamics of BM turnover compared to the hCMEC/D3s, especially considering our prior data on collagen IV deposition by the two cell types. It also matches with existing literature on differences in MMP-9 production between mouse BBB cell types [5].
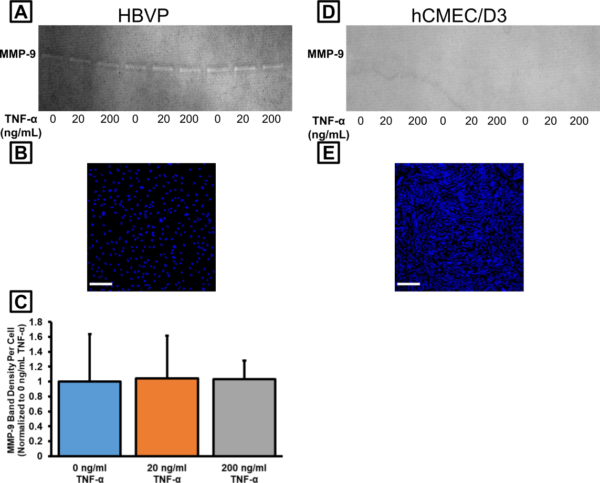
We collected another round of conditioned media after treating with 0, 20, or 200 ng/mL TNF-α for 24 hr. We expected that MMP-9 activity would exhibit a dose-dependent response to treatment. Bands were visible on the gel with our HBVP samples (Fig. 6A), but not on the gel with our hCMEC/D3 samples (Fig. 6D). To better normalize our data, we wanted a rough idea of the number of cells available to produce the MMPs in each sample. We took DAPI-stained images of our cells following sampling of the conditioned media (Fig. 6B,E). We counted the number of cells automatically using ImageJ, averaged the cell density from 3 images for each well, and then estimated the total number of cells in each well. After dividing the band density by the total number of cells, we had an idea of how much “density” was contributed by each individual cell. This was normalized to the 0 ng/mL TNF-α condition. Unexpectedly, there was no difference in MMP-9 activity across our conditions for the HBVPs (Fig. 6C). Either the literature is incorrect (unlikely), or this could be additional evidence that our 1.5 yr-old TNF-α had lost efficacy. In any case, we have recently obtained a fresh lot of human TNF-α for follow-up experiments.
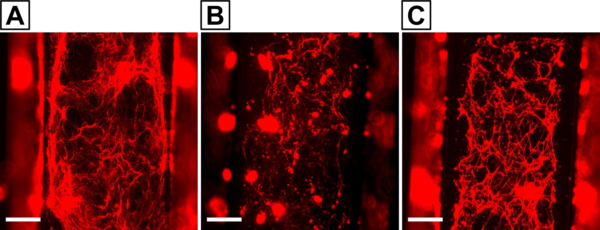
We previously demonstrated an ability to decellularize our µSiM co-cultures with Triton X-100 and ammonia to obtain cell-free BM scaffolds. These may be useful for studying whether the BM influences nanoparticle diffusion across the membrane. In order to examine particles larger than 50 nm, we decided to transition to experiments with mixed porosity (nanopore and 5 µm pore) chips. Preliminary decellularization experiments have yielded unexpected results. Collagen IV deposition on the mixed porosity chips (Fig. 7B,C) seems to be lower than what we observed on purely nanoporous chips (Fig. 7A). Our lab has previously determined that extracellular matrix deposition can be affected by substrate porosity [6], so it is possible that the presence of micropores hampers proper BM formation. We intend to perform a head-to-head comparison between the two membrane types in the near future with more replicates to determine whether or not there is a real, detectable effect.
Conclusions and next steps
We finally have confidence that our cells and BM mimic the orientation of their counterparts in vivo. Our collagen IV deposition data as well as our initial zymograms have indicated that pericytes likely play an important role in BM maintenance and turnover. Unfortunately, we have struggled to recreate TNF-α-mediated increases in MMP-9 activity and the consequential degradation of BM. These issues may be solved after switching to freshly purchased TNF-α.
In order to complete the remaining R61 milestones, we will investigate expression of laminin and fibronectin in our model. In addition to confirming their presence in our in vitro BM, we plan to determine which cells are responsible for their production. This work will be performed using the same hCMEC/D3 and HBVP cell types as before. However, we also plan to conduct BM investigations with iPSC-derived cells. Our first iPSC stain targets will be collagen IV and VE-cadherin, which we hope to image on the Andor Dragonfly Spinning Disc Confocal Microscope to obtain 3D projections similar to figure 1. From there, we may proceed to study the other BM components.
References
[1] X. Cong and W. Kong, “Endothelial tight junctions and their regulatory signaling pathways in vascular homeostasis and disease,” Cellular Signalling, vol. 66, p. 109485, 2020/02/01/ 2020.
[2] C. Leclech, C. F. Natale, and A. I. Barakat, “The basement membrane as a structured surface – role in vascular health and disease,” Journal of Cell Science, vol. 133, p. jcs239889, 2020.
[3] T. Osada, et al., “Interendothelial claudin-5 expression depends on cerebral endothelial cell-matrix adhesion by β(1)-integrins,” Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism, vol. 31, pp. 1972-1985, 2011/10// 2011.
[4] G. A. Rosenberg, E. Y. Estrada, J. E. Dencoff, and W. G. Stetler-Stevenson, “Tumor necrosis factor-α-induced gelatinase B causes delayed opening of the blood-brain barrier: an expanded therapeutic window,” Brain Research, vol. 703, pp. 151-155, 1995/12/12/ 1995.
[5] F. Takata, et al., “Brain pericytes among cells constituting the blood-brain barrier are highly sensitive to tumor necrosis factor-α, releasing matrix metalloproteinase-9 and migrating in vitro,” J Neuroinflammation, vol. 8, p. 106, Aug 2011.
[6] H. H. Chung, S. M. Casillo, S. J. Perry, and T. R. Gaborski, “Porous Substrates Promote Endothelial Migration at the Expense of Fibronectin Fibrillogenesis,” ACS biomaterials science & engineering, vol. 4, pp. 222-230, 2018.