[Rotation] HUVEC Cell Culturing on Aline and SiMPore Devices
Introduction
Aline and SiMPore are producing devices at much higher manufacturing rates than we were capable of in the McGrath lab. In order to verify that these manufactured devices are capable of growing cells and allowing for cell survival, live/dead assays were performed on human umbilical vein endothelial cells (HUVECs) grown 48 hours on either Aline or SiMPore devices. The live/dead assay is frequently used to simultaneously image and quantify the amount of live and dead cells in a culture. The live stain can penetrate cell membranes and is hydrolyzed to produce a green fluorescent compound (measured at Ex/Em = 485/530 nm). The dead stain enters damaged membranes, binds to nucleic acid, and fluoresces red (measured at Ex/Em = 495/635 nm).
Materials and Methods
Culturing Cells
1. Place device in sterilized petri dish and let sit under UV light for 15 minutes to sterilize (Leave lid off the petri dish and SiMPore devices to allow sufficient UV exposure)
2. Add 50 μL of fibronectin to top of membrane (5 μg/cm2 ~ 0.03 mg/ml). Be careful to avoid bubble formation

3. Roll two Kimwipes and generously wet with DI water, spray with ethanol to sterilize, and place around the device inside the petri dish for humidity control
4. Let the fibronectin adhere to the membrane for 1 h at RT (keep covered in dish)
5. Remove fibronectin, rinse with fresh cell culture media
6. Add media to the bottom channel (~ 20 μl). I usually take 50 μl because it is more effective at pushing through the channel and avoiding air bubbles


7. Seed cells as recommended by provider. Please note, available cell seeding area is 5.4 x 5.4 mm = 29.16 mm2. Adjust cell pool density to provide enough cells in 20 μl of media. I seeded HUVECs around 500,000 cells/ml. Top off and exchange media as needed.

8. Cover with lid and incubate at 37°C, 5% CO2, switching media daily (and 2 hours after plating if desired)
9. For optimal phase imaging, add media to form an excess layer on the top of the device. Carefully drop a coverslip atop the media to form a flat interface.
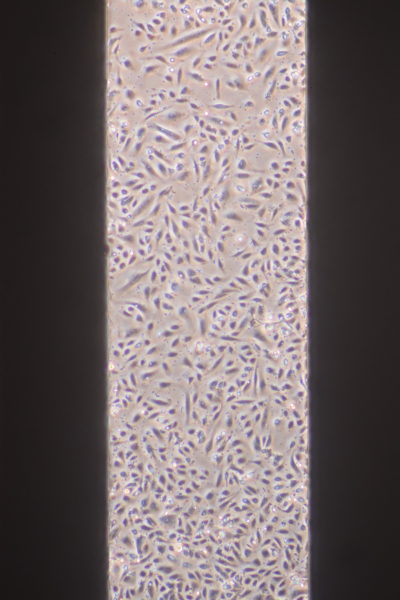
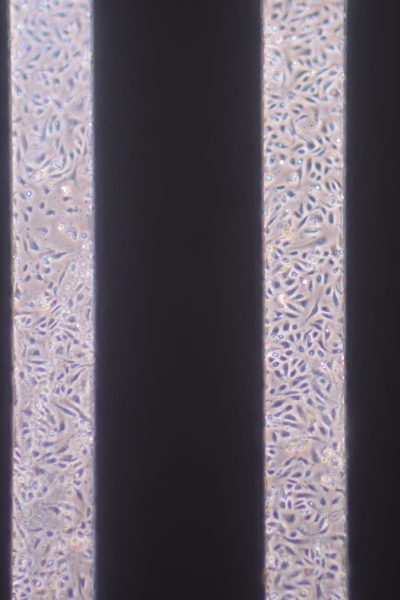
Live/Dead Cell Viability Assay
1. Warm buffer, live stain, and dead stain
2. Add 2 μl Live Stain and 1 μl of Dead Stain to 1 mL Assay Buffer. Mix well
3. Remove media from top channel of device
4. Push 20-50 μl Staining Solution through the bottom channel of the device. Repeat to ensure all media is replaced with Staining Solution
5. Add 25 μl (SiMPore) – 100 μl (Aline) Staining Solution to top channel
6. Cover with lid and incubate at 37°C, 5% CO2 for 15 minutes
7. For imaging, add Staining Solution to form an excess layer on the top of the device. Carefully drop a coverslip atop the solution to form a flat interface. Image immediately with a fluorescent microscope
Results
I quantified the data, counting live and dead cells with >50% of the cell in the image (for edges). Then I divided the total dead by the total cell count for each device type to get the percent dead.
Aline Live/Dead Staining

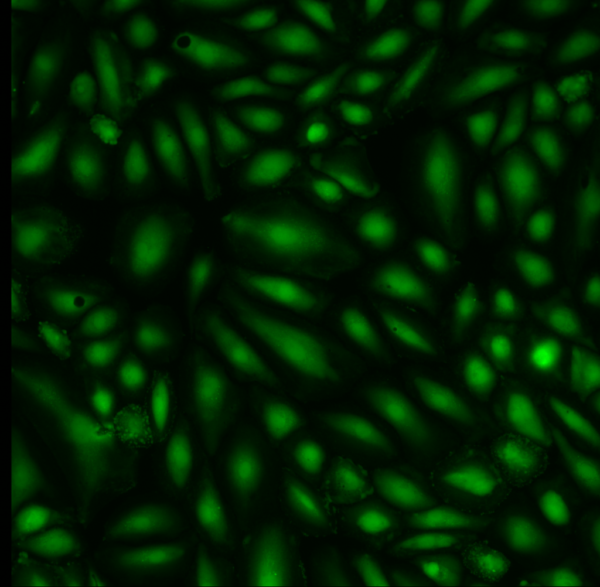

For Aline, I calculated 1.2% dead/98.8% alive.
SiMPore Live/Dead Staining
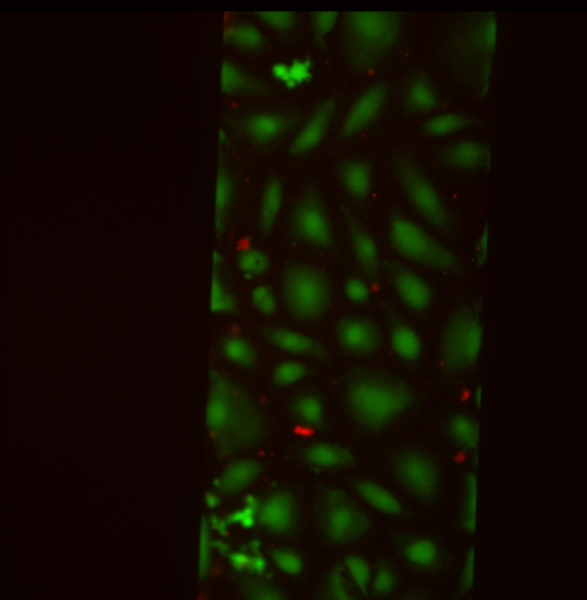
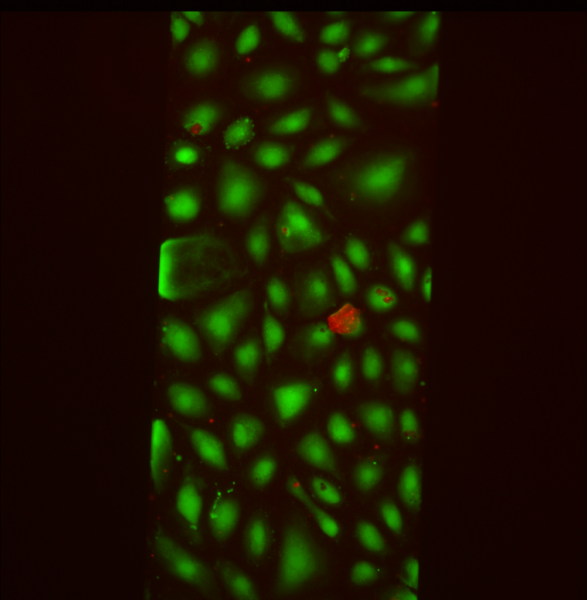
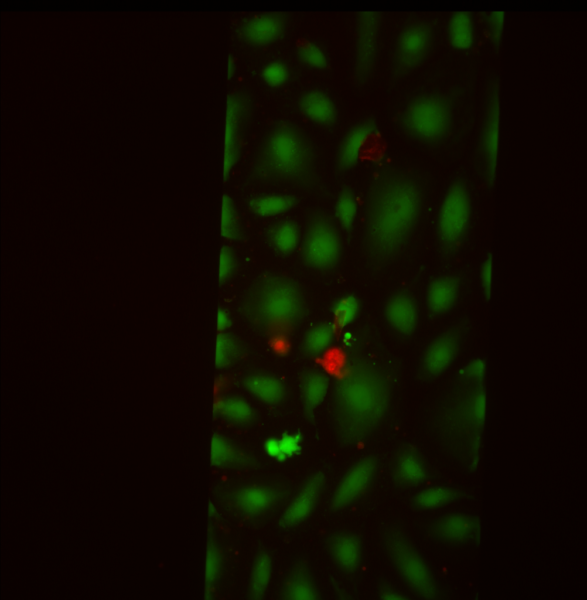
For SiMPore, I calculated 5.8% dead/94.2% alive.
Conclusions
For quantifying the live/dead images, it was hard to tell what was a dead cell and what just debris. I tried to count red dots of decent size and intensity. In addition, it is important to note that for these images, we looked for areas with dead cells, so these percentages are skewed somewhat, and the actual dead percentages are likely lower (aka the devices are better than these numbers imply). Nevertheless, there is still a very high percentage of live cells growing in both types of devices after 48 hours, with the Aline devices appearing to do slightly better than SiMPore.
These are the results from successful cell culturing on the devices. However, there were several unsuccessful attempts to grow cells. Each unsuccessful attempt failed to grow cells in both types of devices, and it appeared that the cells died before settling. We believe the issues are related to fibronectin (either bubble formation over the membrane so fibronectin never contacted the membrane or a reused stock of fibronectin that went bad from repeat freeze/thawing) or potentially from ethanol used to sterilize the KimWipes. It will be important to document cell culturing in these devices well and look for other possible causes of cell death in order to further optimize the protocol and achieve consistency in cell culturing.